


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Kobayashi, Keita; Okumura, Masahiko; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Urata, Shingo*; Suzuya, Kentaro
Scientific Reports (Internet), 13, p.18721_1 - 18721_12, 2023/11
Times Cited Count:1 Percentile:0(Multidisciplinary Sciences)The first sharp peak diffraction peak (FSDP) in the structure factor of amorphous materials is thought to reflect the medium-range order structure in amorphous materials, and the structural origin of the FSDP has been a subject of ongoing debate. In this study, we employed machine learning molecular dynamics (MLMD) with nearly first-principles calculation accuracy to investigate the structural origin of the FSDP in high-density silica glass. First, we successfully reproduced various experimental data of high-density silica glass using MLMD. Furthermore, we revealed that the development (or reduction) of the FSDP in high-density silica glass is characterized by the deformation behavior of ring structures in Si-O covalent bond networks under compression.
Kobayashi, Keita; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Okumura, Masahiko
Materia, 62(3), p.175 - 181, 2023/03
no abstracts in English
Kobayashi, Keita; Okumura, Masahiko; Nakamura, Hiroki; Itakura, Mitsuhiro; Machida, Masahiko; Cooper, M. W. D.*
Scientific Reports (Internet), 12(1), p.9808_1 - 9808_11, 2022/06
Times Cited Count:9 Percentile:71.37(Multidisciplinary Sciences)no abstracts in English
Kobayashi, Keita; Nagai, Yuki; Itakura, Mitsuhiro; Shiga, Motoyuki
Journal of Chemical Physics, 155(3), p.034106_1 - 034106_9, 2021/07
Times Cited Count:5 Percentile:44.89(Chemistry, Physical)no abstracts in English
Kobayashi, Keita; Nakamura, Hiroki; Yamaguchi, Akiko; Itakura, Mitsuhiro; Machida, Masahiko; Okumura, Masahiko
Computational Materials Science, 188, p.110173_1 - 110173_14, 2021/02
Times Cited Count:14 Percentile:73.11(Materials Science, Multidisciplinary)no abstracts in English
Kimoto, Kazushi*; Kawamura, Katsuyuki*; Makino, Hitoshi
Journal of Computer Chemistry, Japan, 19(2), p.46 - 49, 2020/00
This study proposes a 2D coarse-grained molecular dynamics (CGMD) method for the compaction simulation of montmorillonite clay. In the CGMD method, a unit structure of a water-hydrated clay molecule is coarse-grained into a particle. Thus, the deformable molecules are modeled as a set of linearly connected coarse-grained particles. As the inter-particle forces, the intra-molecular bonding and inter-molecular van der Waals forces are considered. For simplicity, the intra-molecular bonding is modeled as a linear harmonic oscillator, while the Lenard-Jones potential is used to define the van der Waals force field. With this model, the mechanical compaction of moistured montmorillonite is numerically simulated to find that 4-6 considerably deformed molecules are layered as a result of the compaction. It is alsofound that the simulated XRD pattern agrees to the experiment in terms of the peak angle.
Machida, Masahiko; Kato, Koichiro*; Shiga, Motoyuki
Journal of Chemical Physics, 148(10), p.102324_1 - 102324_11, 2018/03
Times Cited Count:19 Percentile:70.02(Chemistry, Physical)The isotopologs of liquid water, H O, DO, and TO, are studied systematically by first principles PIMD simulations, in which the whole entity of the electrons and nuclei are treated quantum mechanically. The simulation results are in reasonable agreement with available experimental data on isotope effects, in particular, on the peak shift in the radial distributions of HO and DO and the shift in the evaporation energies. It is found that, due to differences in nuclear quantum effects, the H atoms in the OH bonds more easily access the dissociative region up to the hydrogen bond center than the D (T) atoms in the OD (OT) bonds. The accuracy and limitation in the use of the current density-functional-theory-based first principles PIMD simulations are also discussed. It is argued that the inclusion of the dispersion correction or relevant improvements in the density functionals are required for the quantitative estimation of isotope effects.
O, DO, and TO, are studied systematically by first principles PIMD simulations, in which the whole entity of the electrons and nuclei are treated quantum mechanically. The simulation results are in reasonable agreement with available experimental data on isotope effects, in particular, on the peak shift in the radial distributions of HO and DO and the shift in the evaporation energies. It is found that, due to differences in nuclear quantum effects, the H atoms in the OH bonds more easily access the dissociative region up to the hydrogen bond center than the D (T) atoms in the OD (OT) bonds. The accuracy and limitation in the use of the current density-functional-theory-based first principles PIMD simulations are also discussed. It is argued that the inclusion of the dispersion correction or relevant improvements in the density functionals are required for the quantitative estimation of isotope effects.
Ebihara, Kenichi; Kaburaki, Hideo; Itakura, Mitsuhiro
"Hagane No Kikaiteki Tokusei Ni Oyobosu Suiso No Koka To Sono Hyoka" Shimpojium Yokoshu (USB Flash Drive), 6 Pages, 2014/09
Since hydrogen(H) embrittlement is one factor causing degradation and/or fracture of steel, understanding its mechanism is required. The grain-boundary(GB) decohesion due to segregation of H is considered to cause the delayed fracture of high strength steels and the cold cracking in welding. In the model based on GB decohesion, information of strength of GBs estimated in the atomic scale is used for the estimation of strength or crack propagation in the macroscopic scale. However the modeling between the atomic and the macroscopic scales is not clear. In particular, the validity of the model using the elastic continuum around nano-cracks for stress concentration at the crack tip is not clear. Thus, we examined the difference of the stress distribution around the nano-crack which was estimated by molecular dynamics and by a continuum calculation. As a result, the discrepancy became remarkable at high strain. The stress concentration was not simulated by the elastic continuum model.
Okamoto, Yoshihiro; Madden, P. A.*
Journal of Physics and Chemistry of Solids, 66(1), p.448 - 451, 2005/01
The local structure of molten LaCl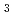 has been characterized by the octahedral coordination (LaCl
has been characterized by the octahedral coordination (LaCl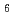 )
)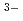 . It is based on X-ray diffraction (XRD) and Raman spectroscopy results. On the other hand, a different structural image was proposed in the neutron diffraction (ND) and the molecular dynamics (MD) studies. These ND and MD works concluded that a coordination behavior changes with cation size in molten rare earth trichlorides. Thus, they reported the coordination number of Cl
. It is based on X-ray diffraction (XRD) and Raman spectroscopy results. On the other hand, a different structural image was proposed in the neutron diffraction (ND) and the molecular dynamics (MD) studies. These ND and MD works concluded that a coordination behavior changes with cation size in molten rare earth trichlorides. Thus, they reported the coordination number of Cl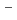 ions around La
ions around La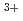 ion is 8.2 in the ND work and 7.9 in the MD work. In the present work, XRD measurements of molten LaCl were performed to investigate the local structure. The coordination number of the 1st La-Cl pair in molten LaCl was 7.1 from analysis of the correlation function G(r). The structural change by increasing anion size was examined by the XRD measurement of molten LaBr. The XRD data were nicely reproduced by the MD simulations with polarizable ionic model.
ion is 8.2 in the ND work and 7.9 in the MD work. In the present work, XRD measurements of molten LaCl were performed to investigate the local structure. The coordination number of the 1st La-Cl pair in molten LaCl was 7.1 from analysis of the correlation function G(r). The structural change by increasing anion size was examined by the XRD measurement of molten LaBr. The XRD data were nicely reproduced by the MD simulations with polarizable ionic model.
Okamoto, Yoshihiro
Nuclear Instruments and Methods in Physics Research A, 526(3), p.572 - 583, 2004/07
Times Cited Count:31 Percentile:86.68(Instruments & Instrumentation)The XAFS functions for highly-disordered materials such as molten salts were simulated by using output data of molecular dynamics calculation. The atomic configuration data obtained from the MD results was directly used as input data in the XAFS simulation code FEFF8. It was assumed that Debye-Waller factor and anharmonic vibration effect could be substituted by an accumulation of the FEFF8 calculations using many atomic configurations. The experimental XAFS functions for platinum, solid and molten salt systems were nicely reproduced by the FEFF8 calculations with 10000-50000 atomic configurations.
Shiga, Motoyuki; Takayanagi, Toshiyuki
Chemical Physics Letters, 378(5-6), p.539 - 547, 2003/09
Times Cited Count:7 Percentile:21.52(Chemistry, Physical)no abstracts in English
Okamoto, Yoshihiro; Akabori, Mitsuo; Ito, Akinori; Ogawa, Toru
Journal of Nuclear Science and Technology, 39(Suppl.3), p.638 - 641, 2002/11
We report local structural features of molten UCl with LiCl-KCl eutectic probed by the U L-edge XAFS(X-ray absorption fine structure). The XAFS measurements were performed in a transmission mode at the BL27B station of the Photon Factory(High Energy Accelerator Organization, Tsukuba, JAPAN). Sample prepared by chlorination of uranium hydride and then reduction with zinc powder was sealed in a quartz cell under reduced pressure. The nearest U-Cl distance and the coordination number of Cl around U ion were obtained by a curve fitting of the 1st shell XAFS function k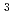
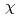 (k). The pair potential in the U-Cl correlation was evaluated from XAFS simulation by combinational use of the MD and the FEFF8. In addition, valence state of uranium in the melt was evaluated by XANES(X-ray absorption near edge structure) spectra.
(k). The pair potential in the U-Cl correlation was evaluated from XAFS simulation by combinational use of the MD and the FEFF8. In addition, valence state of uranium in the melt was evaluated by XANES(X-ray absorption near edge structure) spectra.
Okamoto, Yoshihiro; Fukushima, Kazuko*; Iwadate, Yasuhiko*
Journal of Non-Crystalline Solids, 312(314), p.450 - 453, 2002/10
Local structure of molten ZnBr was investigated by both of the Zn and the Br XAFS (X-ray absorption fine structure) measurements above their K-absorption edge at 723 K. The nearest Zn
was investigated by both of the Zn and the Br XAFS (X-ray absorption fine structure) measurements above their K-absorption edge at 723 K. The nearest Zn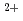 -Br distance was determined to be 2.46
-Br distance was determined to be 2.46 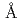 for the Zn XAFS and 2.45 for the Br XAFS. The coordination number of Br ion around Zn was about 4. On the other hand, that of Zn ion around Br was about 2. The 1st Br-Br distance was estimated to be 3.9 . They shows that a tetrahedral coordinate (ZnBr
for the Zn XAFS and 2.45 for the Br XAFS. The coordination number of Br ion around Zn was about 4. On the other hand, that of Zn ion around Br was about 2. The 1st Br-Br distance was estimated to be 3.9 . They shows that a tetrahedral coordinate (ZnBr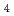 )
)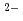 exists and most of the coordinate connects with the next ones through Br ion. The two XAFS functions in the molten ZnBr were reproduced by the combination of the molecular dynamics(MD) simulation and the FEFF7 computations.
exists and most of the coordinate connects with the next ones through Br ion. The two XAFS functions in the molten ZnBr were reproduced by the combination of the molecular dynamics(MD) simulation and the FEFF7 computations.
Shimizu, Futoshi; Kimizuka, Hajime*; Kaburaki, Hideo; Arakawa, Chuichi*
Keisan Kogaku Koenkai Rombunshu, 7(1), p.163 - 166, 2002/05
no abstracts in English
Shimizu, Futoshi; Kimizuka, Hajime*; Kaburaki, Hideo; Arakawa, Chuichi*
Nihon Keisan Kogakkai Rombunshu, 4, p.225 - 230, 2002/04
no abstracts in English
Itakura, Kenichi; Yokokawa, Mitsuo; Shimizu, Futoshi; Kimizuka, Hajime*; Kaburaki, Hideo
Joho Shori Gakkai Kenkyu Hokoku 2001-HPC-88, p.67 - 72, 2001/10
he Earth Simulator which is under development has 640 processor nodes and its peak performance is 40 Tflop/s. In this study, we have evaluated performance of solid molecular dynamics simulation on an SMP node of the Earth Simulator. In molecular dynamics simulation, each particle is influenced by all particles within a cut-off region and the representation of these pairs of particles is made by a matrix. Two matrix representations, compressed row form and jagged diagonal form, are considered for vectorization. The jagged diagonal form is better than the compressed row form in performance on a vector processor for the force calculation of every pairs, because the vector length of the former is longer than that of the latter. However, computational cost for converting the normal matrix form to the jagged diagonal form is quite expensive and the total performance in using the jagged diagonal form is low. Speedup by parallelization with the compressed row form is 2.4 to 2.7 with 8 vector processors.
Fukumoto, Ichiro; Omura, Etsuji*
Seimitsu Kogakkai-Shi, 67(6), p.916 - 921, 2001/06
no abstracts in English
Bastug, T.*; Erkoc, S.*; Hirata, Masaru; Tachimori, Shoichi
Physica E, 8(3), p.223 - 229, 2000/09
Times Cited Count:5 Percentile:27.84(Nanoscience & Nanotechnology)no abstracts in English
Shimizu, Futoshi; Kimizuka, Hajime*; Kaburaki, Hideo; Li, J.*; Yip, S.*
Proceedings of 4th International Conference on Supercomputing in Nuclear Applications (SNA 2000) (CD-ROM), 10 Pages, 2000/09
no abstracts in English
 -
-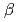 transition of cristobalite, SiO; A Molecular-dynamics study
transition of cristobalite, SiO; A Molecular-dynamics studyKimizuka, Hajime*; Kaburaki, Hideo; Kogure, Yoshiaki*
Physical Review Letters, 84(24), p.5548 - 5551, 2000/06
Times Cited Count:140 Percentile:94.54(Physics, Multidisciplinary)no abstracts in English

